Forcefield parameters and simulations of PEO chains
First and foremost, the CH$_2$ and CH$_3$ are treated as single atoms as we have done when we prepared the initial structure from scratch. Now, let us write the aminoacid.rtp file in the oplsaa.ff folder.
The names of the groups are arbitrary. The group ETF corresponds to the repeat unit at the first end of the chain, whereas the group ETL represents the repeat unit at the final end of the chain. One may notice that in the subsection `bond', the `+' and `-' signs show that whether the units are the first, final or a repeat unit at the middle of the chain. The partial charge for each atom was taken from the TraPPE forcefield. We assume that there is a methyl group at each end of the chain, and its partial atomic charge is 0.
Secondly, in the ffbonded.itp file, one can define all the values for the bond stretching, angle bending as well as torsional potentials. According to TraPPE forcefield, they are as follows:
[ bondtypes ]
; i j func b0 kb
CH2 CH2 1 0.15400 502416.0
CH3 CH2 1 0.15400 502416.0
CH2 O 1 0.14100 502416.0
CH3 O 1 0.14100 502416.0
[ angletypes ]
; i j k func th0 cth
CH2 O CH2 1 112.0 502.166
CH2 CH2 O 1 112.0 418.2
CH2 O CH3 1 112.0 502.166
CH3 CH2 CH2 1 114.0 519.654
[ dihedraltypes ]
; i j k l func coefficients
; OPLS Fourier dihedraltypes translated to Gromacs Ryckaert-Bellemans form
; according to the formula in the Gromacs manual.
CH3 CH2 CH2 O 3 6.98268 -17.73518 0.88694 25.60479 0.00000 0.00000 ;
O CH2 CH2 O 3 8.36779 -25.10337 4.18394 33.47117 0.00000 0.00000 ;
CH3 O CH2 CH2 3 7.94860 -7.892065 2.72284 18.563500 0.00000 0.00000 ;
CH2 CH2 O CH2 3 7.94860 -7.892065 2.72284 18.563500 0.00000 0.00000 ;
CH2 O CH2 CH2 3 7.94860 -7.892065 2.72284 18.563500 0.00000 0.00000 ;
Thirdly, in the ffnonbonded.itp file and the atomtype.itp file, we have to define the parameters for the Lennard-Jones potential as well as the masses and the identity number of the atoms, which are easily written as:
opls_966 CH2 6 14.02700 0.250 A 0.395000 0.382465 ; TraPPE
opls_967 CH3 6 15.03500 0.000 A 0.375000 0.814817 ; TraPPE
opls_968 O 6 15.99940 -0.500 A 0.280000 0.45727
in the atomtypes.atp. Hence, now we can run the command:
gmx pdb2gmx -f initial.pdb
Select 1 for the forcefield if the oplsaa.ff folder is in your working directory, and 8 for not considering the water molecules. A topol.top as well as conf.gro files are then generated. Next is to pack duplicate chains in a box. This can be easily achieved by the command:
which packs 20 PEO chains in a box of size $13\times 13 \times 13~\mathrm{nm^3}$. Note that after execution of this command, last line of the topol.top has to be changed from `Other 1' to `Other 20', indicating that there are in total 20 chain molecules. The energy of the system has to be minimized by the following command:
The file em.mdp contains the input parameters for the energy minimization, which can be found in the folder attached with this document. The run generates a file called em.gro. Similarly, for the equilibriation, we have to run a series of commands that are similar to that of energy minimization:
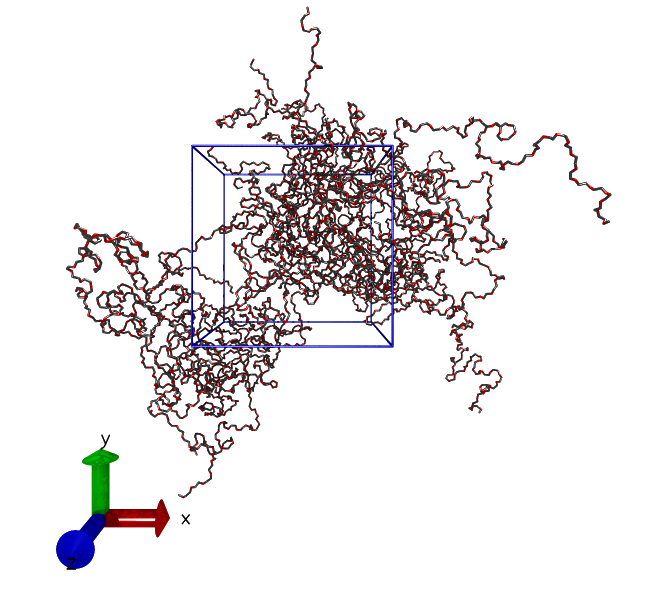
Again, similar to that of the energy minimization process, the run produces a file called md.gro, which is then used as the initial configuration for the production run. During the equilibriation run, the box was compressed to the correct size in a NPT simulation. Figure 1 shows a snapshot of the final configuration of the equilibriation run.
Figure 1: Final configuration of 20 PEO chains each with 302 constituent atoms after the NPT simulation. The purpose of such simulation was to compress the size of the box to a correct value.