MD simulation of system of coarse-grained benzene rings
Here, we are going to simplify the benzene ring by coarse-graining the CH into one unit atom with its specific forcefield parameters. This can be done by modifying several files in the oplsaa.ff folder of the GROMACS package.
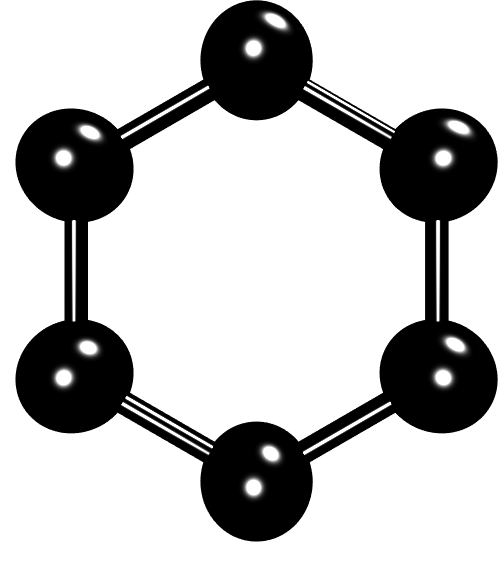
Figure 1: A benzene ring with CH unit coarse-grained as one united atom.
Firstly, let us take a look at the aminoacids.rtp file:
Secondly, a group called [BNN] is created, and the atom type for a CH unit of the benzene ring is defined as opls_971. The bonded and nonbonded forcefield parameters for opls_971 shall be defined. Intuitively, one can know that the bond length and angle for these united atoms must be fixed, meaning a large force constant for the bond stretching and angle bending potentials. Here, we are going to take these values as 800000 and 1000, respectively. The four-body torsional interaction is also fixed at an angle of 180 degrees as the benzene ring is rigid. Hence, the only parameters that we need to think and search for are the ones for the 12-6 Lennard-Jones potential. In that case, we are going to take the parameters from the TraPPE forcefield. In the bonded forcefield file (ffbonded.itp):
[ bondtypes ]
; i j func b0 kb
CHA CHA 1 0.14000 800000.0
[ angletypes ]
; i j k func th0 cth
CHA CHA CHA 1 120.0 1000 ;
In the nonbonded forcefield file (ffnonbonded.itp):
[ atomtypes ]
; full atom descriptions are available in ffoplsaa.atp
; name bond_type mass charge ptype sigma epsilon
opls_971 CHA 6 13.01900 0.000 A 0.369500 0.419875 ; TraPPE
Once these have been modified, one can then move on to creating the initial.pdb file, which can be easily drawn in Avogadros free software. Here is an example of the pdb file:
ATOM 1 C1 BNN 1 0.0 0.0 0.0 1.00 0.00
ATOM 2 C2 BNN 1 0.0 -1.4 0.0 1.00 0.00
ATOM 3 C3 BNN 1 1.21 -2.1 0.0 1.00 0.00
ATOM 4 C4 BNN 1 2.42 -1.4 0.0 1.00 0.00
ATOM 5 C5 BNN 1 2.42 0.0 0.0 1.00 0.00
ATOM 6 C6 BNN 1 1.21 0.7 0.0 1.00 0.00
Thirdly, after these are completed, it can be imported into GROMACS. 156 of the duplicate molecules are then packed into a $2.5\times 2.5\times 2.5$ nm$^3$ box. The GROMACS packing of the duplicate molecules is only roughly done. Unlike PACKMOL, the duplicate molecules can be more precisely packed together. Nonetheless, this imperfection will be fixed by running a short NVT simulation. The following commands should be run:
The last line of the topol.top file must be changed from 1 to 156. The energy minimization and the relaxation in NVT simulation, followed by the production run in NPT simulation. Finally, these can be done by typing the following commands: